Шта је фенилкетонурија
Тамо фенилкетонурија (П.К.У.) то је аутосомно рецесивно наследно метаболичко обољење које погађа 1 од 10.000 особа и чини се да се јавља више у хомозиготности него у хетерозигота.
Припада групи хиперфенилаланинемије, фенилкетонурија значајно угрожава метаболизам фенилаланина, а посебно претварање у тирозин; фенилкетонурија се препознаје по повишеним нивоима фенилаланина и неких деривата у урину (фенилпируват, фенилацетат, фенилактат и фенилацетилглутамин).
Најозбиљнија компликација фенилкетонурије је ментално кашњење.
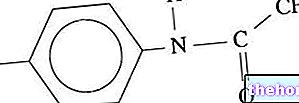
Фенилаланин, тирозин и деривати
Фенилаланин је есенцијална аминокиселина и чини већину протеина у исхрани; може се претворити ензимом фенилаланин хидроксилаза у тирозину (додавањем хидроксилне групе -ОХ). Заузврат, тирозин је прекурсорна аминокиселина за синтезу:
- Л-ДОПА (међупродукт синтезе допамина)
- Епинепхрине
- Норепинефрин (сви неуротрансмитери).

Механизам фенилкетонурије (П.К.У.)
Као што се очекивало, у фенилкетонурији, услед једне или више (укупно 6) хромозомских мутација, експресија (отуда и метаболичка активност) фенилаланин хидроксилазе је практично никаква. Ове промене могу бити различитих врста (од "погрешних" промена до "спајања" дефеката или чак "делимичних брисања"), али оно што је важно је да због ове ензимске неефикасности ниво фенилаланина у крви (који је обично 1 мг / 100 мл) у ДОМИНАНТНОЈ фенилкетонурији лако достижу количине чак 50 пута веће.
Функционисање ензима фенилаланин хидроксилазе: За производњу тирозина (+ дихидробиоптерин), фенилаланин хидроксилази су потребни: фенилаланин, кисеоник и тетрахидробиоптерин (редуковани птеридин који делује као кофактор); реакција је такође реверзибилна и дихидробиоптерин се може поново претворити (захваљујући ензиму дихидроптерин редуктаза) у тетрахидробиоптерину.
Компликације
Фенилкетонурија може изазвати мање или више озбиљне компликације на основу тежине патолошке манифестације и правовремености дијагнозе; Као наследна патологија, фенилкетонурија се разликује по:
- Доминантна, стога окарактерисана ПОТПУНОМ неактивношћу ензима фенилаланин хидроксилазе
- Рецесивна, у којој је активно само 30% укупног ензиматског наслеђа.
Компликације фенилкетонурије могу се приписати и директно пропорционалне метаболичкој акумулацији фенилаланина, његових деривата и смањеној синтези тирозина. У патологији вишак фенилаланина релативно ефикасно филтрира бубрег који га само делимично ресорбује, елиминишући га урином ; међутим, постојаност нивоа хипер-фенилаланинемије одређује метаболичку реакцију молекуларне конверзије у фенилпирувична киселина и / или друге деривате који се лакше исушују (фенилпируват, фенилацетат, фенилактат).
Оно што компликује фенилкетонурију је токсичност фенилаланина, фенилпирувичне киселине и њених деривата према централном нервном систему (ЦНС) .прекомерно присуство у развоју мозга неумитно одређује облик менталне ретардације.
НБ. Концентрације других аминокиселина у плазми су благо смањене, вероватно због повратних информација о интестиналној апсорпцији или реапсорпцији бубрежних тубула.
Оштећење мозга, као озбиљна компликација фенилкетонурије, узроковано је одузимањем других есенцијалних аминокиселина у протеосинтези, посебно у стварању полирибосома, мијелина, норадреналина и серотонина. Фенилкетонурија - није видљива одмах по рођењу, већ након неколико година - ако се не лечи, захтева хоспитализацију детета и потпуно је неповратна.
Напредна фенилкетонурија такође може бити јасно видљива голим оком; високе концентрације фенилаланина, инхибирајући ензим тирозиназа, значајно умањују синтезу меланина смањујући пигментацију коже и косе; надаље, накупљање фенилацетата у коси и кожи даје фенилкетонурицима снажан и непријатан "мирис миша".