Активни састојци: Ранибизумаб
Луцентис 10 мг / мл раствор за ињекције
Улошци за пакет Луцентис доступни су за величине паковања:- Луцентис 10 мг / мл раствор за ињекције
- Луцентис 10 мг / мл раствор за ињекције у напуњеном шприцу
Индикације Зашто се користи Луцентис? За шта је то?
Шта је Луцентис
Луцентис је раствор који се убризгава у око.Луцентис је један из групе лекова који се зову антинеоваскуларни агенси и садржи активну супстанцу звану ранибизумаб.
Чему служи Луцентис?
Луцентис се користи код одраслих за лечење различитих очних обољења која узрокују смањење вида.
Ова стања су резултат оштећења мрежњаче (слоја осетљивог на светлост у задњем делу ока) узрокованог:
- Раст абнормалних крвних судова који испуштају течност (хороидална неоваскуларизација, ЦНВ). То се примећује у стањима као што су старосна макуларна дегенерација (АМД) или патолошка миопија (ПМ).
- Макуларни едем (оток у центру мрежњаче). Ово отицање може бити узроковано дијабетесом (стање које се назива дијабетички едем макуле (ДМЕ)) или зачепљењем вена мрежњаче (стање које се назива оклузија ретиналне вене (РВО)).
Како Луцентис ради
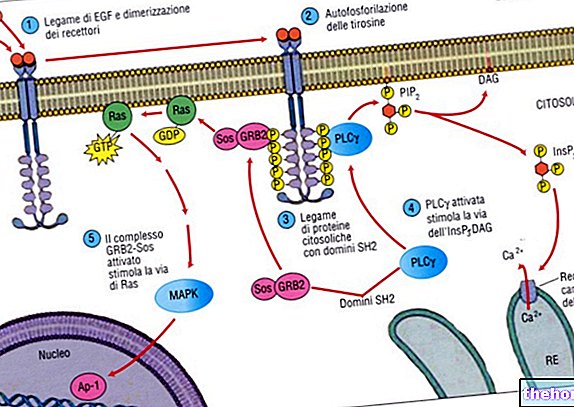
Луцентис посебно препознаје и веже протеин који се налази у оку, назван људски ендотелни васкуларни фактор раста А (ВЕГФ-А). Када је вишак, ВЕГФ-А изазива абнормални раст крвних судова и отицање у оку што може довести до смањења крвних судова вид у условима као што су АМД, ПМ, ДМЕ или РВО. Везујући ВЕГФ-А, Луцентис може блокирати његово деловање и спречити абнормални раст и отицање.
У овим условима, Луцентис може помоћи у стабилизацији и у многим случајевима побољшати вид.
Контраиндикације Када се Луцентис не сме користити
Не смете да примате Луцентис
- ако сте алергични на ранибизумаб или неки други састојак овог лека
- ако имате „инфекцију на једном оку или у околини.
- ако имате бол или црвенило (тешка интраокуларна упала) на једном оку.
Предострожности при употреби Шта треба да знате пре него што узмете лек Луцентис
Пре него што примите Луцентис, разговарајте са својим лекаром.
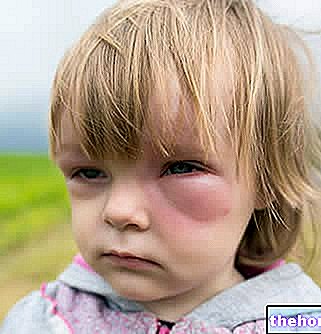
- Луцентис се даје као "ињекција" у око.Повремено се може појавити „инфекција у унутрашњем делу ока“, бол или црвенило (упала), одвајање или пуцање једног од слојева у задњем делу ока (одвајање мрежњаче или пуцање и одвајање или пуцање епитела) након третман са Луцентисом. ретинални пигмент), или замућење сочива (катаракта). Важно је да се што пре идентификује и лечи "инфекција или одвајање мрежњаче. Одмах обавестите свог лекара ако осетите било какве знаке као што су бол у очима или појачана нелагодност, погоршање црвених очију", замагљен или смањен вид, повећање телесних ћелија у виду или повећаној осетљивости на светлост.
- Код неких пацијената, притисак у оку може се повећати за кратко време одмах након ињекције. Овај догађај је нешто што можда нећете приметити, па би ваш лекар требало да провери након сваке ињекције.
- Реците свом лекару ако сте раније имали очне проблеме или третмане ока, или сте имали мождани удар или знакове пролазних исхемијских напада (слабост или парализа удова или лица, потешкоће у говору или разумевању). Ове информације ће се узети у обзир приликом процене да ли је Луцентис одговарајући третман за вас.
Деца и адолесценти (млађи од 18 година)
Употреба Луцентиса код деце и адолесцената није проучавана и стога се не препоручује.
Интеракције Који лекови или храна могу променити ефекат Луцентиса
Реците свом лекару ако користите, недавно сте користили или бисте могли да користите било који други лек.
Упозорења Важно је знати да:
Трудноћа и дојење
- Женама у репродуктивном периоду треба саветовати да током лечења користе ефикасну контрацепцију.
- Нема искуства са употребом лека Луцентис код трудница, па потенцијални ризици нису познати. Ако сте трудни, сумњате или планирате трудноћу, молимо вас да о томе разговарате са својим лекаром пре почетка терапије леком Луцентис.
- Не препоручује се употреба Луцентиса током дојења јер није познато да ли се Луцентис излучује у мајчино млеко. Питајте свог лекара или фармацеута за савет пре него што почнете да узимате лек Луцентис.
Вожња и управљање машинама
Након третмана Луцентисом може доћи до привременог замућења вида. Ако се то догоди, немојте управљати возилима и машинама док се ово стање не реши.
Доза, начин и време примене Како се користи Луцентис: Дозирање
Како ће вам Луцентис бити дат
Ваш офталмолог Луцентис даје као једну ињекцију у око под локалном анестезијом. Уобичајена доза једне ињекције је 0,05 мл (која садржи 0,5 мг активног састојка). Интервал између две дозе убризгане у исто око треба да буде најмање четири недеље. Све ињекције ће вам дати ваш очни лекар.
Пре ињекције, лекар ће вам темељито очистити око како би спречио инфекцију, а лекар ће вам дати и локални анестетик како би умањио или спречио бол који може настати приликом ињекције.
Лечење је почело једном ињекцијом Луцентиса месечно. Лекар ће пратити стање ока и на основу одговора на лечење одлучити да ли је и када потребно даље лечење.
Детаљна упутства за корисника могу се пронаћи на крају овог упутства под насловом "Како припремити и применити Луцентис".
Старији пацијенти (65 година и више)
Луцентис се може користити за пацијенте старије од 65 година без прилагођавања дозе.
Ако сте пропустили дозу Луцентиса
Контактирајте свог лекара или болницу што је пре могуће како бисте заказали термин.
Пре него што прекинете лечење Луцентисом
Ако размишљате о прекиду лечења Луцентисом, посетите следећу посету и разговарајте о томе са својим лекаром. Ваш лекар ће вас посаветовати и одлучити колико дуго ћете морати да се лечите Луцентисом.
Ако имате додатних питања о употреби овог лека, обратите се свом лекару.
Нежељени ефекти Који су нежељени ефекти Луцентиса
Као и сви лекови, и овај лек може изазвати нежељена дејства, мада се она не морају јавити код свих.
Нуспојаве повезане са давањем Луцентиса су последица самог лека и поступка убризгавања и углавном утичу на око.
Најозбиљнији нежељени ефекти описани су у наставку:
Уобичајени озбиљни нежељени ефекти (могу се јавити у до 1 на 10 пацијената): одвајање или суза у задњем делу ока (одвајање или пуцање мрежњаче), манифестовано бљесковима светлости са пловцима до привременог смањења вида или замућењем вида сочива (катаракта). Мање чести озбиљни нежељени ефекти (могу се јавити у до 1 на 100 пацијената): слепило, инфекција очне јабучице (ендофталмитис) са упалом унутар ока.
Симптоми које можете осетити описани су у одељку 2 ове брошуре (прочитајте одељак 2 "Шта треба да знате пре него што вам буде даван лек Луцентис"). Одмах се обратите лекару ако се појави било који од ових нежељених ефеката.
У наставку су описани најчешће пријављени нежељени ефекти:
Веома чести нежељени ефекти (могу се јавити код више од 1 на 10 пацијената)
Визуелни споредни ефекти укључују: упалу ока, крварење у стражњем делу ока (ретинално крварење), сметње вида, бол у очима, честице или мрље у виду (флоатери), локализовано црвенило очију, иритацију ока, осећај тела у страној материји око, повећана производња суза, упала или инфекција ивице капака, суво око, црвено или сврбеж ока и повећан притисак у оку.
Невизуелни споредни ефекти укључују: грлобољу, зачепљен нос, цурење из носа, главобољу и болове у зглобовима.
Остали нежељени ефекти који се могу јавити након терапије леком Луцентис описани су у наставку:
Уобичајени нежељени ефекти
Визуелни нежељени ефекти укључују: смањену оштрину вида, отицање дела ока (увеа, рожњача), упалу рожњаче (предњи део ока), мале трагове на површини ока, замагљен вид, крварење на месту убризгавања, крварење у оку, свраб из ока, црвенило и отицање (коњунктивитис), осетљивост на светлост, нелагодност у очима, отицање капака, бол у очним капцима. Невизуелни споредни ефекти укључују: инфекцију уринарног тракта, смањење црвених крвних зрнаца (са симптомима као што су као што су умор, недостатак ваздуха, вртоглавица, бледило), анксиозност, кашаљ, мучнина, алергијске реакције попут осипа, осипа, свраба и црвенила коже.
Ретки нежељени ефекти
Визуелни нежељени ефекти укључују: упалу и крварење у предњем делу ока, сакупљање гноја у оку, промене у централном делу очне површине, бол или иритацију на месту убризгавања, ненормалан осећај у оку, иритацију капака.
Ако добијете било које нежељено дејство, обратите се свом лекару, што укључује и све могуће нуспојаве које нису наведене у овом упутству.
Пријављивање нежељених ефеката
Ако добијете било које нежељено дејство, обратите се свом лекару, што укључује и све могуће нуспојаве које нису наведене у овом упутству. Такође можете пријавити нежељена дејства директно путем националног система за пријављивање наведеног у Додатку В. Пријављивањем нежељених ефеката можете помоћи у пружању више информација о безбедности овог лека.
Истек и задржавање
- Чувајте овај лек ван погледа и дохвата деце.
- Немојте користити овај лек након истека рока ваљаности наведеног на кутији и на бочици иза ознаке „Рок употребе“ и „Рок употребе“.
- Чувати у фрижидеру (2 ° Ц - 8 ° Ц). Немојте замрзавати.
- Бочицу чувајте у спољном паковању ради заштите лека од светлости.
- Не користите пакет који је оштећен.
Садржај паковања и друге информације
Шта Луцентис садржи
- Активна супстанца је ранибизумаб (10 мг / мл). Сваки мл садржи 10 мг ранибизумаба.
- Помоћни састојци су α, α-трехалоза дихидрат; хистидин хидрохлорид, монохидрат; хистидин; полисорбат 20; вода за ињекције.
Опис како Луцентис изгледа и садржај паковања
Луцентис је раствор за ињекције у бочици (0,23 мл). Раствор је водени, бистар, безбојан до бледо жут.
Луцентис се испоручује у паковању које садржи стаклену бочицу ранибизумаба са чепом од хлоробутил гуме, тупу иглу за филтрирање (18Г к 1½ ″, 1,2 мм к 40 мм, 5 микрометара) за извлачење садржаја бочице, иглу за ињекцију (30Г к ½ ″, 0,3 мм к 13 мм) и шприц (1 мл) за извлачење садржаја бочице за интравитреалну ињекцију.Све компоненте су само за једнократну употребу.
Следеће информације су намењене само здравственим радницима: Такође погледајте одељак "Како ће вам бити додељен Луцентис".
Како припремити и применити Луцентис
Бочице за једнократну употребу само за интравитреалну примену Луцентиса треба да даје квалификовани офталмолог са искуством у интравитреалним ињекцијама.
Код влажног АМД -а, оштећења вида услијед ДМЕ -а, макуларног едема секундарног РВО -а или ЦНВ -а секундарног ПМ -а, препоручена доза Луцентиса је 0,5 мг у једној интравитреалној ињекцији.
Ово одговара убризганој запремини од 0,05 мл. Интервал између две дозе убризгане у исто око треба да буде најмање четири недеље.
Лечење почиње једном ињекцијом месечно све док се не постигне максимална оштрина вида и / или нема знакова активности болести као што су промене видне оштрине и промене у другим знацима и симптомима болести током наставка лечења. Пацијенти са мокром АМД, ДМЕ и РВО, можда ће бити потребно започети терапију са три или више узастопних месечних ињекција.
Према томе, лекар треба да одлучи о интервалима праћења и лечења и да се заснива на активности болести, што се утврђује проценом оштрине вида и / или анатомских параметара.
Ако, по мишљењу лекара, оштрина вида и анатомски параметри указују на то да пацијент нема користи од наставка лечења, Луцентис треба прекинути.
Праћење активности болести може укључивати клинички преглед, функционалне процене или технике снимања (попут оптичке кохерентне томографије или флуоресцеинске ангиографије).
Ако се пацијенти лече према режиму лечења и продужења, након постизања максималне оштрине вида и / или у одсуству знакова активности болести, интервали лечења могу се постепено продужавати све до знакова болести или до погоршања вида функција. Интервал између третмана треба постепено продужавати за највише две недеље код пацијената са влажном АМД, а може се продужити до месец дана код пацијената са ДМЕ. Интервали третмана се такође могу постепено продужавати у лечењу РВО, али не и тамо је довољан податак за утврђивање трајања ових интервала. Након поновног појављивања активности болести, интервал третмана треба сходно томе скратити.
У лечењу оштећења вида узрокованог ЦНВ -ом секундарним након ПМ, многим пацијентима је потребна само једна или две ињекције током прве године, док некима је потребно чешће лечење.
Луцентис и ласерска фотокоагулација у ДМЕ и макуларни едем секундарни према БРВО
Постоји одређено искуство да се Луцентис примењује истовремено са ласерском фотокоагулацијом. Када се користи истог дана, Луцентис треба применити најмање 30 минута након ласерске фотокоагулације. Луцентис се може давати пацијентима који су претходно били на ласерској фотокоагулацији.
Луцентис и фотодинамичка терапија са Висудине -ом у ЦНВ секундарно у односу на ПМ
Нема искуства са применом Луцентиса у комбинацији са Висудинеом. Пре примене Луцентиса треба визуелно проверити на присуство честица и промену боје.
Поступак убризгавања треба извести под асептичним условима, који укључују хируршку дезинфекцију руку, стерилне рукавице, стерилну завесу и стерилни блефаростат (или еквивалент), и могућност извођења стерилне парацентезе (ако је потребно). Извођење интравитреалне процедуре, пацијентове анамнезу треба пажљиво проценити за реакције преосетљивости. У складу са клиничком праксом, пре ињекције треба применити одговарајућу анестезију и локални антимикробни лек широког спектра за дезинфекцију периокуларне, очне и капне површине.
Да бисте припремили Луцентис за интравитреалну ињекцију, следите доле наведена упутства:
Све компоненте су стерилне и само за једнократну употребу. Не смије се користити било која компонента на чијем паковању постоје знаци оштећења или неовлаштеног рада. Не може се гарантовати стерилност ако печат на амбалажи компоненте није нетакнут.
- Дезинфикујте спољни део гуменог чепа бочице пре сакупљања.
- Асептично причврстите иглу филтера од 5 µм (18Г к 1½ ″, 1,2 мм к 40 мм, 5 µм, испоручено) на штрцаљку од 1 мл (испоручено). Уметните тупу иглу филтера у средину поклопца док не додирне дно бочица.
- Извуците сву течност из бочице држећи је у усправном положају, благо нагнутом како бисте олакшали потпуно повлачење.
- Уверите се да је клип шприца повучен довољно уназад приликом пражњења бочице да бисте потпуно испразнили иглу филтера.
- Оставите иглу филтера тупу у бочици и извадите шприц из ње. Баците филтер иглу након што извучете садржај бочице и немојте је користити за интравитреалну ињекцију.
- Чврсто и асептично поставите ињекциону иглу (30Г к ½ ″, 0,3 мм к 13 мм, испоручена) на шприц.
- Пажљиво уклоните поклопац са игле за ињекцију без одвајања ињекционе игле са шприца. Напомена: Држите жуту подлогу ињекционе игле док уклањате поклопац.
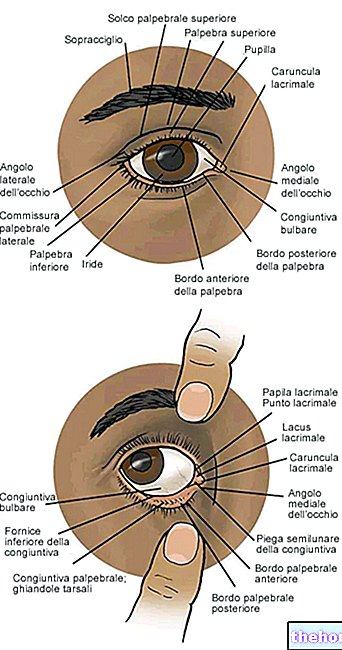
- Пажљиво избаците ваздух из шприца и подесите дозу на 0,05 мл означену на шприцу Шприца је спремна за ињекцију. Напомена: Немојте чистити иглу за убризгавање. Немојте повлачити клип уназад. Убаците иглу за убризгавање 3,5-4,0 мм напред према лимбусу, у стаклену комору, избегавајући хоризонтални меридијан и усмеравајући иглу према средини глобуса. запремина ињекције 0,05 мл; промените склерално место за накнадне ињекције. Након ињекције, немојте покривати иглу нити је одвајати од шприца. Одложите употребљени шприц заједно са иглом у одговарајући контејнер или у складу са локалним захтевима.
Упутство о извору: АИФА (Италијанска агенција за лекове). Садржај објављен у јануару 2016. Присутне информације можда нису ажурне.
Да бисте имали приступ најновијој верзији, препоручљиво је да приступите веб страници АИФА (Италијанска агенција за лекове). Одрицање од одговорности и корисне информације.
01.0 НАЗИВ ЛИЈЕКА
ЛУЦЕНТИС 10 МГ / МЛ раствор за убризгавање
02.0 КВАЛИТАТИВНИ И КВАНТИТАТИВНИ САСТАВ
Један мл садржи 10 мг ранибизумаба *. Свака бочица садржи 2,3 мг ранибизумаба у 0,23 мл раствора.
* Ранибизумаб је фрагмент хуманизованог моноклоналног антитела произведен у ћелијама Есцхерицхиа цоли технологијом рекомбинантне ДНК.
За потпуну листу помоћних супстанци погледајте одељак 6.1.
03.0 ФАРМАЦЕУТСКИ ОБЛИК
Раствор за ињекције
Бистри, безбојни до бледо жути водени раствор.
04.0 КЛИНИЧКЕ ИНФОРМАЦИЈЕ
04.1 Терапијске индикације
Луцентис је индициран код одраслих за:
• Лечење старосне неоваскуларне (влажне) макуларне дегенерације (АМД)
• Лечење оштећења вида узрокованог дијабетичким едемом макуле (ДМЕ)
• Лечење оштећења вида узрокованог „макуларним едемом који је последица оклузије вене мрежњаче (грана РВО или централни РВО)
• Лечење оштећења вида узроковано хороидалном неоваскуларизацијом (ЦНВ) секундарно патолошкој кратковидости (ПМ)
04.2 Дозирање и начин примене
Луцентис треба да даје квалификовани офталмолог са искуством у интравитреалним ињекцијама.
Дозирање за лечење мокрог АМД
Препоручена доза Луцентиса је 0,5 мг која се даје месечно као једна интравитреална ињекција. Ово одговара убризганој запремини од 0,05 мл.
Третман се примењује месечно и наставља се док се не постигне максимална оштрина вида, тј. Оштрина вида пацијента је стабилна за три узастопне месечне контроле које се раде током лечења ранибизумабом.
Због тога је потребно месечно пратити оштрину вида пацијената.
Третман треба наставити ако праћење укаже на смањење видне оштрине услед влажног АМД -а. Затим би требало применити месечне ињекције док се поново не постигне стабилна оштрина вида за три узастопне месечне контроле (то подразумева најмање две ињекције). Интервал између две дозе не сме бити мањи од месец дана.
Дозирање за лечење оштећења вида узрокованог ДМЕ -ом или макуларним едемом који је последица РВО -а
Препоручена доза Луцентиса је 0,5 мг која се даје месечно као једна интравитреална ињекција. Ово одговара убризганој запремини од 0,05 мл.
Третман се примењује месечно и наставља се док се не постигне максимална оштрина вида, тј. Оштрина вида пацијента је стабилна за три узастопне месечне контроле које се раде током лечења ранибизумабом. Ако нема побољшања видне оштрине током прве три ињекције, не препоручује се наставак лечења.
Због тога је потребно месечно пратити оштрину вида пацијената.
Третман треба наставити када праћење укаже на смањену оштрину вида услед ДМЕ -а или макуларног едема који је последица РВО -а. Затим би требало применити месечне ињекције све док се поново не постигне стабилна оштрина вида за три месечне контроле. Узастопно (ово укључује најмање две ињекције). Интервал између две дозе не би требало да буде мањи од месец дана.
Луцентис и ласерска фотокоагулација у ДМЕ и макуларни едем секундарни према БРВО
Постоји одређено искуство у примени Луцентиса истовремено са ласерском фотокоагулацијом (видети одељак 5.1). Када се даје истог дана, Луцентис треба дати најмање 30 минута након ласерске фотокоагулације. Луцентис се може дати пацијентима који су претходно примили ласерску фотокоагулацију.
Дозирање за лечење оштећења вида узрокованог ЦНВ -ом секундарно у односу на ПМ
Лечење треба започети једном ињекцијом.
Ако се праћењем открију знаци активности болести, као што је смањена оштрина вида и / или знаци повреде, препоручује се даље лечење.
Праћење болести може укључивати клинички преглед, оптичку кохерентну томографију (ОЦТ) или флуоресцеинску ангиографију (ФА).
Док је неким пацијентима можда потребна само једна или две ињекције током прве године лечења, некима ће можда бити потребно чешће лечење (видети одељак 5.1). Због тога се препоручује месечно праћење током прва два месеца, а најмање свака три месеца током прве године лечења. Након прве године, лекар може одредити учесталост праћења.
Интервал између две дозе не сме бити мањи од месец дана.
Луцентис и фотодинамичка терапија са Висудине -ом у ЦНВ секундарно у односу на ПМ
Нема искуства са применом Луцентиса у комбинацији са Висудинеом.
Посебне популације
Хепатична инсуфицијенција
Луцентис није проучаван код пацијената са инсуфицијенцијом јетре. Међутим, за овај став нису потребна посебна разматрања.
Инсуфицијенција бубрега
Није потребно прилагођавање дозе код пацијената са бубрежном инсуфицијенцијом (видети одељак 5.2).
Старији грађани
Код старијих особа није потребно прилагођавање дозе. Постоји „ограничено“ искуство код пацијената са ДМЕ старијим од 75 година.
Педијатријска популација
Безбедност и ефикасност лека Луцентис код деце и адолесцената млађих од 18 година нису утврђени. Нема доступних података.
Начин примене
Бочице за једнократну употребу само за интравитреалну употребу.
Пре примене Луцентиса треба визуелно проверити на присуство честица и промену боје.
Поступак за ињекцију мора се извести под асептичним условима, који укључују дезинфекцију руку као и за било који хируршки захват, стерилне рукавице, стерилну завесу и стерилни блефаростат (или еквивалент) и могућност извођења стерилне парацентезе (ако је потребно анамнезу реакција преосетљивости пацијента треба пажљиво проценити пре интравитреалне процедуре (видети одељак 4.4). У складу са клиничком праксом, пре ињекције треба применити одговарајућу анестезију и локални антимикробни лек широког спектра за дезинфекцију периокуларне, очне и капне површине.
За информације о припреми Луцентиса погледајте одељак 6.6.
Ињекциону иглу убаците 3,5-4,0 мм позади лимбуса, у комору стакластог тела, избегавајући хоризонтални меридијан и усмеравајући иглу према центру очне јабучице. Убризгати ињекциону запремину од 0,05 мл; променити склерално место за наредне ињекције.
04.3 Контраиндикације
Преосетљивост на активну супстанцу или било коју помоћну супстанцу наведену у одељку 6.1.
Пацијенти са тренутним или сумњивим окуларним или периокуларним инфекцијама.
Пацијенти са сталном тешком интраокуларном упалом.
04.4 Посебна упозорења и одговарајуће мере опреза при употреби
Реакције повезане са интравитреалном ињекцијом
Интравитреалне ињекције, укључујући оне са Луцентисом, биле су повезане са ендофталмитисом, интраокуларном упалом, регматогеним одвајањем мрежњаче, руптуром мрежњаче и јатрогеном трауматском катарактом (видети одељак 4.8). За примену Луцентиса увек треба користити одговарајуће технике асептичког убризгавања. Осим тога, пацијенте треба пратити недељу дана након ињекције како би се омогућило брзо лечење у случају инфекције. Пацијенте треба упутити како да без одлагања пријаве све симптоме који указују на ендофталмитис или било који од горе наведених догађаја.
Повећање очног притиска
У року од 60 минута од ињекције Луцентиса примећено је пролазно повећање очног притиска (ИОП). Такође је примећено продужено повећање интраокуларног притиска (видети одељак 4.8). Интраокуларни притисак и перфузију главе оптичког нерва треба пратити и на одговарајући начин лечити.
Билатерални третман
Ограничени подаци о билатералној употреби Луцентиса (укључујући дозирање истог дана) не указују на повећан ризик од системских нежељених догађаја у поређењу са унилатералним третманом.
Имуногеност
Са Луцентисом постоји потенцијал за имуногеност. С обзиром да постоји могућност повећане системске изложености код испитаника са ДМЕ, не може се искључити повећани ризик од развоја преосетљивости у овој популацији пацијената. Пацијенте такође треба едуковати о томе како пријавити ако се интраокуларна упала погорша јер би то могао бити клинички симптом који се може приписати стварање интраокуларних антитела.
Истовремена употреба са другим анти-ВЕГФ (васкуларни ендотелни фактор раста)
Луцентис се не сме примењивати истовремено са другим анти-ВЕГФ лековима (системским или окуларним).
Луцентис прекид
Не треба давати дозу и не треба наставити лечење пре следећег заказаног третмана у случају:
• смањење најбоље кориговане оштрине вида (БЦВА) ≥30 слова у односу на последњу процену;
• интраокуларни притисак ≥30 ммХг;
• прекид мрежњаче;
• "субретинално крварење које се протеже до центра фовее, или ако је опсег крварења ≥50% укупне површине лезије";
• интраокуларна операција изведена или планирана у претходних или наредних 28 дана.
Пуцање пигментног епитела ретине
Фактори ризика повезани са настанком руптуре пигментног епитела ретине након анти-ВЕГФ терапије за влажни АМД укључују велико и / или високо одвајање пигментног епитела ретине. Приликом започињања терапије Луцентисом, потребан је опрез код пацијената са овим факторима ризика за пуцање ретиналног пигментног епитела.
Регматогено одвајање мрежњаче или макуларне рупе
Лечење треба прекинути код особа са регматогеним одвајањем мрежњаче или макуларним рупама стадија 3 или 4.
Становништво са ограниченим подацима
Постоји само ограничено искуство у лечењу субјеката са ДМЕ секундарним у односу на дијабетес типа И. Луцентис није проучаван код пацијената који су претходно примали интравитреалне ињекције, код пацијената са активним системским инфекцијама, пролиферативном дијабетичком ретинопатијом или код пацијената са истовременим медицинским стањима као што је одвајање мрежњаче или макуларна рупа. Такође нема искуства у лечењу Луцентисом код пацијената са дијабетесом са ХбАлц већим од 12% и неконтролисаном хипертензијом. Лекар треба да узме у обзир недостатак информација при лечењу ових пацијената.
Код пацијената са ПМ, постоје ограничени подаци о ефекту Луцентиса код пацијената који су претходно лечени неуспешном фотодинамичком терапијом вертепорфином (вПДТ). Надаље, иако је констатован ефекат забележен код особа са субфовеалним и јукстафовеалним лезијама, нема довољно података о ефекту Луцентиса код ПМ субјеката са екстрафовеалним лезијама.
Системски ефекти након интравитреалне примене
Системски нежељени догађаји, укључујући неокуларне крварења и артеријске тромбоемболијске догађаје, пријављени су након интравитреалне ињекције инхибитора ВЕГФ.
Постоје ограничени подаци о безбедности лечења ДМЕ -а, макуларног едема узрокованог РВО -ом и ЦНВ -ом секундарно у односу на ПМ код пацијената са историјом можданог удара или пролазних исхемијских напада. Посебан опрез је потребан при лечењу таквих пацијената (видети одељак 4.8).
Претходне епизоде РВО, исхемијске гране и централног РВО
Постоји ограничено искуство у лечењу пацијената са претходним епизодама РВО и пацијената са исхемијском граном РВО (БРВО) и централним РВО (ЦРВО). Код пацијената са РВО који имају губитак видне функције са клиничким знацима иреверзибилне исхемије, лечење је Не препоручује се.
04.5 Интеракције са другим лековима и други облици интеракција
Нису спроведена конвенционална испитивања интеракција.
За комбиновану примену фотодинамичке терапије (ПДТ) са вертепорфином и Луцентисом у влажној АМД и ПМ, видети одељак 5.1.
За комбиновану употребу ласерске фотокоагулације и Луцентиса у лечењу ДМЕ и БРВО, видети одељке 4.2 и 5.1.
04.6 Трудноћа и дојење
Жене у репродуктивном периоду / контрацепција код жена
Жене у репродуктивном периоду треба да користе ефикасну контрацепцију током лечења.
Трудноћа
За ранибизумаб нису доступни клинички подаци о изложености трудноћи. Студије на мајмунима циномолгус нису показале директне или индиректне штетне ефекте на трудноћу или ембрионални / фетални развој (видети одељак 5.3). Системска изложеност ранибизумабу је ниска након очне примене, али због механизма деловања ранибизумаб треба сматрати потенцијално тератогеним и ембрио / фетотоксичним. Због тога се ранибизумаб не сме примењивати током трудноће, осим ако очекиване користи надмашују потенцијални ризик за фетус. Женама које планирају трудноћу и које су лечене ранибизумабом препоручује се да сачекају најмање 3 месеца након последње дозе ранибизумаба пре зачећа бебе.
Трудноћа
Није познато да ли се Луцентис излучује у мајчино млеко. Препоручује се да не дојите док користите Луцентис.
Плодност
Нема доступних података о плодности.
04.7 Утицај на способност управљања возилима и машинама
Поступак лечења Луцентисом може изазвати пролазне сметње вида које могу утицати на способност управљања возилима и рада са машинама (видети одељак 4.8). Пацијенти који имају ове симптоме не би требали возити или управљати машинама док ови пролазни поремећаји вида не престану.
04.8 Нежељени ефекти
Сажетак сигурносног профила
Већина нежељених реакција пријављених након примене Луцентиса повезана је са поступком интравитреалне ињекције.
Најчешће пријављене очне нуспојаве након ињекције Луцентиса су: бол у оку, хиперемија ока, повишен интраокуларни притисак, витреитис, одвајање стакластог ткива, крварење у мрежњачи, сметње вида, пловци (крварење у стакластом тијелу), иритација ока, осјећај страног тијела око, повећано сузење, блефаритис, суво око и сврбеж ока.
Најчешће пријављене неокуларне нежељене реакције су главобоља, назофарингитис и артралгија.
Мање често пријављене, али озбиљније нежељене реакције укључују ендофталмитис, слепило, одвајање мрежњаче, руптуру мрежњаче и јатрогену трауматску катаракту (видети одељак 4.4).
Пацијенте треба обавестити о симптомима ових потенцијалних нежељених реакција и упутити их да обавесте свог лекара ако осете знакове као што су бол у очима или повећана нелагодност, погоршање црвенила ока, замагљен или смањен вид, повећан број стакластих лелујача или " повећана осетљивост на светлост.
Нежељене реакције пријављене након примене Луцентиса у клиничким студијама сажете су у доњој табели.
Табела нежељених реакција #
Нежељене реакције су наведене према органским системима и учесталости према следећој конвенцији: врло честе (≥1 / 10), честе (≥1 / 100,
Инфекције и инфестације
Веома честа Назофарингитис
заједнички Инфекција уринарног тракта *
Поремећаји крви и лимфног система
заједнички Анемија
Поремећаји имунолошког система
заједнички Преосетљивост
Психијатријски поремећаји
заједнички Анксиозност
Поремећаји нервног система
Веома честа Главобоља
Поремећаји ока
Веома честа Витреитис, одвајање стакластог тела, крварење у мрежњачи, сметње вида, бол у очима, флоатери у стакластом тијелу, крварење у коњуктиви, иритација ока, осећај страног тела у оку, повећана лакримација, блефаритис, суво око, очна хиперемија, сврбеж ока.
заједнички Дегенерација мрежњаче, поремећаји мрежњаче, одвајање мрежњаче, кидање мрежњаче, одвајање пигментног епитела ретине, пуцање пигментног епитела ретине, смањена оштрина вида, крварење у стакластом телу, поремећаји стакластог тела, увеитис, иритис, иридоциклитис, катаракта, поткапсуларна катаракта постериорна капсула кератитис, абразија рожњаче, реакција предње коморе, замагљен вид, крварење на месту убризгавања, крварење у очима, коњунктивитис, коњунктивитис
алергијски, очни исцједак, свјетлосни бљескови, фотофобија, очна нелагодност, едем капака, бол у очним капцима, хиперемија коњунктиве.
необичан Слепоћа, ендофталмитис, хипопион, хифема, кератопатија, синехије шаренице, наслаге рожњаче, едем рожњаче, стријеве рожњаче, бол на месту убризгавања, иритација на месту убризгавања, абнормални осећај у оку, иритација капака.
Поремећаји дисања, грудног коша и медијастинума
заједнички Кашаљ
Гастроинтестинални поремећаји
заједнички Мучнина
Поремећаји коже и поткожног ткива
заједнички Алергијске реакције (осип, осип, свраб, еритем)
Поремећаји мишићно -коштаног система и везивног ткива
Веома честа Артралгија
Дијагностички тестови
Веома честа Повећан интраокуларни притисак
# Нежељене реакције су дефинисане као нежељени догађаји (код најмање 0,5 процентних поена пацијената) који су се јавили са већом стопом (најмање 2 процентна поена) код пацијената који су били на терапији Луцентисом 0,5 мг у поређењу са онима који су примали контролни третман (лажни или ПДТ вертепорфин).
* забележено само у популацији са ДМЕ
Нежељене реакције повезане са категоријом лекова
У студијама ИИИ са влажним АМД-ом, укупна учесталост неокуларних крварења, нежељених догађаја потенцијално повезаних са инхибиторима ВЕГФ-а (фактор раста ендотелних судова), била је благо повећана код пацијената лечених ранибизумабом. Међутим, не постоји константан образац између различитих крварења. Постоји теоретски ризик од артеријских тромбоемболијских догађаја, укључујући мождани удар и инфаркт миокарда, који су резултат интравитреалне употребе инхибитора ВЕГФ. Ниска учесталост артеријских тромбоемболијских догађаја примећена је у клиничким испитивањима са Луцентисом код пацијената са АМД, ДМЕ, РВО и ПМ и нису уочене разлике међу групама ранибизумаба у поређењу са контролом.
Пријављивање сумње на нежељене реакције
Извештавање о сумњама на нежељене реакције које се јављају након добијања дозволе за лек важно је јер омогућава континуирано праћење односа користи и ризика од лека. Здравствени радници се моле да пријаве сваку сумњу на нежељену реакцију преко Италијанске агенције за лекове. , веб страница: хттпс://ввв.аифа.гов.ит/цонтент/сегналазиони-реазиони-авверсе.
04.9 Предозирање
Случајеви случајног предозирања забележени су из клиничких испитивања на мокром АМД-у и постмаркетиншких података. Нежељене реакције које су најчешће биле повезане са овим случајевима биле су повишен очни притисак, пролазно слепило, смањена оштрина вида, едем рожњаче и бол. Ако дође до предозирања, интраокуларни притисак треба пратити и лекар сматрати неопходним.
05.0 ФАРМАКОЛОШКА СВОЈСТВА
05.1 Фармакодинамичка својства
Фармакотерапијска група: Офталмолози, анти-неоваскуларни агенси, АТЦ ознака: С01ЛА04
Ранибизумаб је хуманизовани рекомбинантни фрагмент моноклоналног антитела усмерен против људског васкуларног ендотелног фактора раста А (ВЕГФ-А).Везује се са високим афинитетом за изоформе ВЕГФ-А (нпр. ВЕГФ110, ВЕГФ121 и ВЕГФ165), спречавајући тако везивање ВЕГФ-А за његове рецепторе ВЕГФР-1 и ВЕГФР-2. За његове рецепторе доводи до пролиферације ендотелних ћелија на неоваскуларизацију и повећање васкуларне пропустљивости за које се сматра да доприносе прогресији неоваскуларног облика старосне макуларне дегенерације, патолошке миопије или смањеног вида узрокованог или дијабетичким макуларним едемом или „Макуларним едемом секундарним у односу на РВО.
Третман влажног АМД -а
За влажну АМД, безбедност и клиничка ефикасност Луцентиса процењиване су у три 24-месечне рандомизоване, двоструко слепе, лажне или активно контролисане студије код пацијената са неоваскуларном АМД. Укупно 1.323 пацијената (879 лечених и 444 контроле) било је укључено у ове студије.
У студији ФВФ2598г (МАРИНА), 716 пацијената са минимално класичним или окултним лезијама хороидалне неоваскуларизације (ЦНВ) без класичне компоненте примило је месечне интравитреалне ињекције Луцентиса 0,3 мг (н = 238) или 0,5 мг (н = 240) или лажне ињекције (н = 238).
У студији ФВФ2587г (АНЦХОР), 423 пацијента са претежно класичним ЦНВ -ом примило је један од следећих третмана: 1) месечне интравитреалне ињекције Луцентиса 0,3 мг и лажног ПДТ -а (н = 140); 2) месечне интравитреалне ињекције Луцентиса 0,5 мг и лажног ПДТ -а (н = 140); или 3) интравитреалне лажне ињекције и ПДТ са вертепорфином (н = 143). ПДТ са вертепорфином или лажним је даван заједно са почетном ињекцијом Луцентиса, а затим свака 3 месеца ако је флуорангиографија показала постојаност или наставак васкуларног цурења.
Кључни налази сажети су у табелама 1, 2 и на слици 1.
Табела 1 Резултати у 12. и 24. месецу у студији ФВФ2598г (МАРИНА)
ап
Табела 2 Резултати у 12. и 24. месецу у студији ФВФ2587г (СИДРА)
Сто
Резултати обе студије показали су да би наставак лечења ранибизумабом такође могао бити од користи код пацијената који су изгубили ≥15 слова најбоље кориговане оштрине вида (БЦВА) у првој години лечења.
Студија ФВФ3192г (ПИЕР) је рандомизирана, двоструко слепа, лажно контролисана студија осмишљена да процени безбедност и ефикасност Луцентиса код 184 пацијената са свим облицима неоваскуларне АМД. Пацијенти су примили интравитреалне ињекције Луцентиса 0,3 мг (н = 60) или 0,5 мг (н = 61) или лажне ињекције (н = 63) једном месечно у 3 узастопне дозе, након чега следи једна доза која се даје једном у 3 месеца. Од 14. месеца студије, пацијенти лечени лажном ињекцијом примани су лечење ранибизумабом, а од 19. месеца могли су да се изводе чешћи третмани. Пацијенти лечени Луцентисом у ПИЕР студији примили су у просеку укупно 10 третмана.
Примарна крајња тачка ефикасности била је средња промена видне оштрине након 12 месеци у поређењу са основном вредношћу. Након почетног повећања видне оштрине (након месечне дозе), у просеку се видна оштрина пацијената смањивала са тромесечним дозирањем, враћајући се на почетну вредност у 12. месецу, а овај ефекат је задржан код већине лечених пацијената. Са ранибизумабом (82%) Месец 24. Подаци ограниченог броја испитаника који су преведени на лечење ранибизумабом након више од годину дана лажног лечења сугеришу да рано започињање лечења може бити повезано са бољим задржавањем „видне оштрине“.
И у студијама МАРИНА и у АНЦХОР-у побољшање видне оштрине примећено са Луцентисом од 0,5 мг након 12 месеци било је праћено користима које су пријавили пацијенти мерене резултатом Упитника за визуелне функције Националног института за очи (ВФК-25). Разлике између Луцентиса 0,5 мг и две контролне групе су процењене са вредностима п у распону од 0,009 до
Ефикасност Луцентиса у лечењу влажног АМД-а потврђена је у постмаркетиншким студијама АМД-а. Подаци из две студије (МОНТ БЛАНЦ, БПД952А2308 и ДЕНАЛИ, БПД952А2309) нису показали додатне ефекте комбиноване примене вертепорфина (Висудине ПДТ) и Луцентис у поређењу са самим Луцентисом.
Лечење оштећења вида услед ДМЕ
Безбедност и ефикасност лека Луцентис процењиване су у две рандомизиране, двоструко слепе, лажно контролисане или активне 12-месечне студије код пацијената са смањеним видом због дијабетичког макуларног едема. Укупно су ове студије биле укључене. Од 496 пацијената (336 активних и 160 контрола), већина је имала дијабетес типа ИИ, 28 лечених пацијената имало је дијабетес типа И.
У другој фази студије Д2201 (РЕСОЛВЕ), 151 пацијент је лечен ранибизумабом (6 мг / мл, н = 51, 10 мг / мл, н = 51) или лажним (н = 49) са једном "интравитреалном ињекцијом месечно. док се не испуне унапред дефинисани критеријуми. Почетна доза ранибизумаба (0,3 мг или 0,5 мг) могла се удвостручити у било ком тренутку током студије након прве ињекције. Ласерска фотокоагулација је била дозвољена као спасоносни третман од 3. месеца у обе руке. Студија имао је два дела: истраживачки део (првих 42 пацијената посећено у 6. месецу) и потврдни део (преосталих 109 пацијената посећено у 12. месецу).
Кључни налази из потврдног дела студије (2/3 пацијената) сажети су у Табели 3.
Табела 3 Исходи у 12. месецу у студији Д2201 (РЕШИ) (укупна популација студије)
ап
У студији ИИИ фазе Д2301 (РЕСТОРЕ), 345 пацијената са оштећењем вида због макуларног едема било је рандомизовано да прими или „интравитреалну ињекцију од 0,5 мг ранибизумаба као монотерапију и ласерску лажну фотокоагулацију (н = 116), или комбинацију од 0,5 мг ранибизумаб и ласерска фотокоагулација (н = 118) или „лажна ињекција и ласерска фотокоагулација (н = 111). Лечење ранибизумабом започето је месечним интравитреалним ињекцијама и настављало се док оштрина вида није остала стабилна најмање три узастопне месечне провере. Лечење је настављено када је примећено смањење БЦВА услед прогресије ДМЕ. Ласерска фотокоагулација је примењена на почетку истог дана, најмање 30 минута пре ињекције ранибизумаба, а након тога према потреби на основу критеријума ЕТДРС.
Кључни налази сажети су у табели 4 и на слици 2.
Табела 4 Резултати у 12. месецу у студији Д2301 (ОБНОВИ)
ап
Ефекат је био доследан у већини подгрупа, међутим, субјекти са прилично високим БЦВА на почетку (> 73 слова) са макуларним едемом и централном дебљином мрежњаче
Побољшање видне оштрине у 12. месецу примећено са Луцентисом 0,5 мг било је пропраћено користима главних функција повезаних са видом које је пријавио пацијент, мерено резултатом Упитника за визуелне функције Националног института за очи (ВФК-25). Не постоје разлике због лечења Разлика између Луцентиса 0,5 мг и контролне групе је процењена са п-вредношћу од 0,0137 (ранибизумаб моно) и 0,0041 (ранибизумаб + ласер) за композитни резултат ВФК-25.
У обе студије, визуелно побољшање пратило је континуирано смањење макуларног едема мерено као централна дебљина мрежњаче (ЦРТ).
Третман оштећења вида узрокованог макуларним едемом који је последица РВО
Клиничка безбедност и ефикасност Луцентиса код пацијената са оштећењем вида услед макуларног едема који је последица РВО-а процењивани су у рандомизованим, двоструко слепим, контролисаним испитивањима: БРАВО и ЦРУИСЕ који су регрутовали пацијенте са БРВО (н = 397) и ЦРВО (н = 392) У обе студије, пацијенти су примили или 0,3 мг или 0,5 мг ранибизумаба интравитреално или лажне ињекције. Након 6 месеци, пацијенти у лажној контролној руци премештени су у групу ранибизумаба 0,5 мг. У БРАВО студији, ласерска фотокоагулација као третман за спасавање био дозвољен у свим оружјима од 3. месеца.
Кључни налази из студија БРАВО и ЦРУИСЕ приказани су у Табелама 5 и 6
Табела 5 Резултати у 6. и 12. месецу (БРАВО)
ап
Табела 6 Резултати у 6. и 12. месецу (КРСТАРЕЊЕ)
ап
У обе студије, визуелно побољшање пратило је континуирано и значајно смањење макуларног едема мерено у смислу дебљине централне ретине.
Код пацијената са БРВО -ом (студија БРАВО и продужење студије ХОРИЗОН): Након 2 године, пацијенти који су били лечени лажним ињекцијама у првих 6 месеци, а затим прешли на лечење ранибизумабом, имали су повећање АВ (& симп; 15 слова) упоредиво са тим пацијената који су били на лечењу ранибизумабом од почетка студије (& симп; 16 слова). Међутим, број пацијената који су навршили 2 године био је ограничен и у студији ХОРИЗОН су заказане само тромјесечне посете. Постоји довољно доказа да се закључе препорукама о када треба започети лечење ранибизумабом код пацијената са БРВО.
Код пацијената са ЦРВО (студија ЦРУИСЕ и продужење студије ХОРИЗОН): После 2 године, пацијенти који су у првих 6 месеци били лечени лажним ињекцијама, а затим преведени на терапију ранибизумабом, нису показали повећање АВ (& симп; 6 слова) у поређењу са онима пацијената који су лечени ранибизумабом од почетка студије (& симп; 12 слова).
Побољшање видне оштрине примећено током лечења ранибизумабом у 6. и 12. месецу праћено је добробитима које су пријавили пацијенти мерене анкетним упитником за визуелне функције Националног института за очи (НЕИ ВФК-25) за блиске и удаљене активности Разлика између Луцентиса 0,5 мг и утврђено је да се контролна група налази између вредности п између 0,02 и 0,0002.
Лечење оштећења вида услед ЦНВ -а секундарно са ПМ
Безбедност и клиничка ефикасност Луцентиса код пацијената са оштећењем вида услед ЦНВ у ПМ потврђени су на основу 12-месечних података из кључне рандомизоване, двоструко слепе, контролисане студије Ф2301 (РАДИАНЦЕ). Ова студија је имала за циљ да процени два различита режима дозирања. ранибизумаба 0,5 мг примењеног интравитреалном ињекцијом у односу на вертепорфински ПДТ (вПДТ, визудинска фотодинамичка терапија). 277 пацијената је рандомизовано у једну од следећих група:
• Група И (ранибизумаб 0,5 мг, режим лечења одређен критеријумима "стабилности" дефинисаним као нема промене БЦВА у односу на процене у претходна два месеца).
• Група ИИ (ранибизумаб 0,5 мг, режим лечења одређен према критеријуму "активности болести" дефинисан као оштећење вида приписује се интра- или субретиналној течности или активном истицању изазваном лезијама ЦНВ-а, што доказују ОЦТ и / или АФ).
• Група ИИИ (пацијенти лечени вПДТ - са могућношћу лечења ранибизумабом почевши од 3. месеца).
Током 12 месеци студије, пацијенти су примили у просеку 4,6 ињекција (опсег 1-11) у Групи И и 3,5 ињекције (опсег 1-12) у Групи ИИ. Међу пацијентима који припадају групи ИИ, која одражава препоручену дозу (видети одељак 4.2), 50,9% пацијената је прошло терапију са 1 до 2 ињекције, 34,5% 3 до 5 ињекција и 14,7% је дало 6 до 12 ињекција током 12-месечне студије . 62,9% пацијената ИИ групе није захтевало ињекције током других 6 месеци студије.
Кључни налази РАДИАНЦЕ -а сажети су у табели 7 и на слици 5.
Табела 7 Резултати у 3. и 12. месецу (РАДИАНЦЕ)
ап
б Упоредна контрола до 3. месеца Пацијенти рандомизовани да приме вПДТ били су подобни за лечење ранибизумабом 3. месеца (у ИИИ групи, 38 пацијената је добило ранибизумаб 3. месеца)
Побољшање вида било је праћено смањењем дебљине централне ретине.
У поређењу са групом која је примала вПДТ, пацијенти у групама леченим ранибизумабом пријавили су корист (п-вредност
Педијатријска популација
Безбедност и ефикасност ранибизумаба код деце још нису утврђени.
Европска агенција за лекове одустала је од обавезе достављања резултата студија са Луцентисом у свим подскупинама педијатријске популације за неоваскуларну АМД, оштећење вида услед ДМЕ, оштећење вида услед секундарног макуларног едема због РВО и оштећење вида услед секундарног ЦНВ ПМ (видети одељак 4.2 за информације о педијатријској употреби).
05.2 Фармакокинетичка својства
Након месечне интравитреалне примене Луцентиса код пацијената са неоваскуларном АМД, серумске концентрације ранибизумаба су генерално биле ниске, са максималним нивоима (Цмак) генерално испод концентрације ранибизумаба потребне за инхибирање биолошке активности ВЕГФ-а за 50% (11-27 нг / мЛ) , оцењено на тесту ин витро пролиферација ћелија). Цмак је била пропорционална дози у целом распону доза од 0,05 до 1,0 мг / око. Код ограниченог броја пацијената са ДМЕ -ом, откривене концентрације у серуму указују на то да се не може искључити нешто већа системска изложеност онима које су примећене код пацијената са неоваскуларном АМД. Серумске концентрације ранибизумаба код пацијената са РВО биле су сличне или незнатно веће од оних које су примећене код пацијената са неоваскуларном АМД.
На основу популационе фармакокинетичке анализе и серумског клиренса ранибизумаба за неоваскуларне АМД пацијенте лечене дозом од 0,5 мг, средњи полувреме елиминације ранибизумаба у стакластом тијелу је приближно 9 дана. У време месечне интравитреалне примене Луцентиса 0,5 мг / око, очекује се да ће серумски Ц ранибизумаба, достигнут приближно 1 дан након дозе, генерално кретати између 0,79 и 2,90 нг / мл, док се очекује да Цмин генерално варира између 0,07 и 0,49 нг / мл. Процењује се да су концентрације ранибизумаба у серуму приближно 90.000 пута ниже од концентрација у стакластом телу.
Пацијенти са бубрежном инсуфицијенцијом: Нису спроведене конвенционалне студије за испитивање фармакокинетике Луцентиса код пацијената са бубрежном инсуфицијенцијом. У „фармакокинетичкој анализи у популацији пацијената са неоваскуларном АМД, 68% (136 од 200) пацијената имало је„ бубрежну инсуфицијенцију (46,5% блага [50-80 мЛ / мин], 20% умерена [30 -50 мЛ / мин] и 15% озбиљан [системски клиренс је био нешто нижи, али то није било клинички значајно.
Пацијенти са инсуфицијенцијом јетре: Нису спроведене конвенционалне студије које би испитивале фармакокинетику Луцентиса код пацијената са инсуфицијенцијом јетре.
05.3 Предклинички подаци о безбедности
Билатерална интравитреална примена ранибизумаба мајмунима циномолгус у дозама између 0,25 мг / око и 2,0 мг / око једном у 2 недеље до 26 недеља довела је до очних ефеката зависних од дозе.
Интраокуларно, повећање брзине и ћелија зависило је од дозе у предњој комори, а врхунац је достигао 2 дана након ињекције. Озбиљност упалног одговора опћенито се смањује с накнадним ињекцијама или током периода опоравка. које су такође имале тенденцију да зависе од дозе и опстајале су до краја периода лечења.У студији од 26 недеља, озбиљност упале стакластог тела се повећавала са бројем ињекција. Међутим, реверзибилност је примећена након периода опоравка. Природа и трајање упале задњег сегмента указују на имунолошки посредовани одговор антитела, који може бити клинички ирелевантан. Формирање катаракте је примећено код неких животиња након релативно дугог периода интензивне упале, што сугерише да су промене сочива биле секундарне до тешке упале. Након примене интравитреалних ињекција примећено је пролазно повећање очног притиска, без обзира на дозу.
Микроскопске очне промене биле су повезане са упалом и нису указивале на дегенеративне процесе.Упале грануломатозне промене забележене су на оптичком диску неких очију. Ове промене задњег сегмента су се смањиле, а у неким случајевима и решиле, током периода опоравка.
Није било знакова системске токсичности након интравитреалне примене. Серумска и стакласта антитела на ранибизумаб нађена су у подскупу третираних животиња.
Нису доступни подаци о канцерогености или мутагености.
Код трудних мајмуна, интравитреална ињекција ранибизумаба резултирала је максималном системском изложеношћу 0,9-7 пута најгоре клиничке изложености није узроковала развојну токсичност или тератогеност, те није имала утјецаја на тежину или структуру тијела. тератоген и ембрио / фетотоксичан на основу фармаколошког дејства.
Одсуство посредованих ефеката ранибизумаба на развој ембриона / фетуса је вероватно повезано углавном са немогућношћу фрагмента Фаб да пређе плаценту. Међутим, описан је случај са високим нивоом ранибизумаба у мајчином серуму и присуством ранибизумаба у феталном серуму, што сугерише да је антитело против ранибизумаба деловало као протеин (који садржи ФЦ регију) који транспортује ранибизумаб, чиме се смањује његово уклањање из мајчиног серума и дозвољавајући његово преношење у плаценту. Будући да су тестови на развој ембрија / фетуса проведени на здравим трудним животињама и да неке болести (попут дијабетеса) могу промијенити плацентну пропусност за Фаб фрагмент, студију је потребно тумачити с опрезом.
06.0 ФАРМАЦЕУТСКЕ ИНФОРМАЦИЈЕ
06.1 Помоћне супстанце
α, α-трехалоза дихидрат
Хистидин хидрохлорид, монохидрат
Хистидин
Полисорбат 20
Вода за ињекције
06.2 Некомпатибилност
У недостатку студија компатибилности, овај лек се не сме мешати са другим лековима.
06.3 Период важења
3 године
06.4 Посебне мере предострожности при складиштењу
Чувати у фрижидеру (2 ° Ц - 8 ° Ц).
Немојте замрзавати.
Бочицу чувајте у спољном паковању ради заштите лека од светлости.
06.5 Природа непосредног паковања и садржај паковања
0,23 мл стерилног раствора у бочици (стакло типа И) са чепом (хлоробутил гума), 1 тупом иглом за филтрирање (18Г к 1½ ", 1,2 мм к 40 мм, 5 мцм), 1 иглом за ињекцију (30Г к ½", 0,3 мм к 13 мм) и 1 шприц (полипропилен) (1 мл). Паковање садржи 1 бочицу.
06.6 Упутства за употребу и руковање
Бочица, игла за ињекцију, игла за филтер и шприц су само за једнократну употребу. Поновна употреба може изазвати инфекцију или другу болест / повреду. Све компоненте су стерилне. Не смије се користити било која компонента на чијем паковању постоје знаци оштећења или неовлаштеног рада. Не може се гарантовати стерилност ако печат на амбалажи компоненте није нетакнут.
Да бисте припремили Луцентис за интравитреалну ињекцију, следите доле наведена упутства:
1. Пре сакупљања дезинфикујте спољни део гуменог чепа бочице.
2. Асептично причврстите иглу филтера од 5 мцм (18Г к 1½ ", 1,2 мм к 40 мм, испоручено) на шприц од 1 мл (испоручено).Уметните тупу иглу филтера у средину поклопца док не додирне дно бочице.
3. Извуците сву течност из бочице држећи је у усправном положају, благо нагнутом да бисте олакшали потпуно повлачење.
4. Уверите се да је клип шприца повучен довољно уназад приликом пражњења бочице да бисте потпуно испразнили иглу филтера.
5. Оставите иглу филтера тупу у бочици и извадите шприц из ње. Одбаците иглу филтера након што извучете садржај бочице и немојте је користити за интравитреалну ињекцију.
6. Чврсто и асептично причврстите ињекциону иглу (30Г к ½ ", 0,3 мм к 13 мм, испоручено) на шприц.
7. Пажљиво уклоните поклопац са ињекционе игле без одвајања ињекционе игле са шприца.
Напомена: Држите жуту подлогу ињекционе игле док уклањате поклопац.
8. Пажљиво избаците ваздух из шприца и подесите дозу на 0,05 мл означену на шприцу Шприца је спремна за ињекцију.
Напомена: Не чистите ињекциону иглу и не повлачите клип уназад.
Након ињекције, немојте покривати иглу нити је одвајати од шприца. Одложите употребљени шприц заједно са иглом у одговарајући контејнер или у складу са локалним захтевима.
07.0 НОСИЛАЦ ОВЛАШЋЕЊА ЗА ПРОМЕТ
Новартис Еуропхарм Лимитед
Вимблехурст Роад
Хорсхам
Западни Сасекс, РХ12 5АБ
УК
08.0 БРОЈ ОДЛИКЕ ЗА ПРОМЕТ
ЕУ/1/06/374/001
037608027
09.0 ДАТУМ ПРВОГ ОДОБРЕЊА ИЛИ ОБНОВЕ ОВЛАШЋЕЊА
Датум прве ауторизације: 22. јануар 2007
Датум последњег обнављања: 24. јануар 2012
10.0 ДАТУМ РЕВИЗИЈЕ ТЕКСТА
05/2014








-cloruro.jpg)